

Во второй половине прошлого столетия на гомогенный процесс жидкофазного аэробного окисления возлагались очень большие надежды, как на относительно дешевый способ получения кислородсодержащих веществ из углеводородного сырья. Однако данный способ оказался низкоселективным по многим ценным промежуточным продуктам (гидропероксиды, спирты, альдегиды) и интерес к нему исследователей во всем мире несколько снизился, тем более что практическая реализация процесса сопровождалась рядом трудностей, связанных с разделением продуктов, выделением и регенерацией гомогенных катализаторов. Тем не менее по всему миру ряд крупнотоннажных производств оказались конкурентноспособными, в частности, процессы, связанные с окислением циклогексана, этилбензола, кумола и его производных с получением соответствующих гидропероксидов, которые находят широкое применение в различных областях химической промышленности. Как правило, эти процессы в настоящее время проводятся в отсутствие катализаторов с инициаторами радикально-цепных схем окисления. К сожалению, в большинстве случаев инициированное окисление характеризуется достаточно низкими скоростью протекания реакции и конверсией исходного сырья.
Несмотря на значительный практический опыт эксплуатации этих производств, а также выполнение огромного количества научно-исследовательских работ, загадки гидропероксидного жидкофазного окисления и поныне разгаданы далеко не полностью и остаются одними из главных и труднорешаемых для специалистов, работающих в этой области.
Большинство исследований, направленных на усовершенствование процессов окисления углеводородов до гидропероксидов, связано с использованием солей металлов переменной валентности (Сo, Mn, Cu, Ni и др.) в качестве катализаторов. Однако данные каталитические системы не нашли практического применения, так как наряду с повышением скорости окисления в их присутствии наблюдается преждевременный нецелевой распад гидропероксида. Кроме этого, в присутствии этих катализаторов приходится поддерживать низкую конверсию исходного углеводорода, поскольку с ростом концентрации гидропероксида в оксидате резко возрастает и скорость его разложения. С позиции устранения подобных недостатков практический и научный интерес представляет разработка технологии так называемого «органокатализа», которая может быть применима для аэробного окисления различного углеводородного сырья.
Более двадцати лет назад наметились принципиально новые тенденции в развитии процессов жидкофазного окисления, связанные с применением в качестве катализатора органических соединений – N-гидроксифталимида (N-ГФИ) и его аналогов. Это соединение, впервые полученное в XIX веке прусским химиком Ласаром Коном, было названо фталилгидроксиламином [1]. Только через столетие N-ГФИ впервые будет использован в качестве катализатора процесса взаимодействия эфиров с диэтилазодикарбоксилами [2], а Я. Ишией, М. Масуи, Р. Шелдоном, Ф. Рекуперо и др. будет продолжено исследование каталитических свойств данного вещества уже в реакциях окисления химических соединений: алкиларенов, алкенов, алкинов, спиртов, эфиров, аминов, амидов, силанов до различных кислородсодержащих продуктов [3–6]. Соединение, помимо этого, может быть использовано для повышения цетанового числа бензина [7], а также проявляет специфические антимикробные свойства в отношении микроорганизмов, резистентных к другим антибиотикам [8]. Производные N-ГФИ являются промежуточными продуктами в получении антикоррозионных агентов [9], функционально замещенных альдегидов [10]. Функционализация C–H-связей становится значимым процессом в синтезе и модификации сложных органических молекул, являющихся лекарственными веществами, компонентами агрохимикатов и мономерами для синтеза полимеров [11, 12]. Производные N-ГФИ могут применяться и для C–O, C–N функционализации, например, в получении π-аллилпалладиевых комплексов, реакциях радикального каскада и в модификации лекарств и природных веществ [13].
Таким образом, уникальное соединение становится объектом интереса многих исследователей при поиске новых путей проведения селективных химических превращений. N-гидроксифталимид является высокоэффективным с технологической точки зрения катализатором-инициатором, способствующим интенсивному, но селективному и стабильному окислению различных органических субстратов; его применение может дать явные экономические и экологические преимущества. Но, несмотря на достаточно широкую применимость этого уникального вещества и интерес к инновационной и гибкой технологии органокатализа, фталимидные катализаторы не были масштабно внедрены в производство.
Для промышленной реализации технологии органокатализа необходимо проведение всесторонних исследований фталимидного окисления и накопление фундаментально-теоретической базы. С этой точки зрения определенный интерес представляет поиск решения проблем, связанных с ограниченной растворимостью N-ГФИ в углеводородах. В научно-технической литературе описаны два пути, позволяющие устранить указанный недостаток и осуществлять процесс в гомогенной среде. Первое направление связано с поиском полярного сорастворителя [14] с учетом того, что он должен быть инертным в условиях окисления ко всем компонентам реакционной массы, хорошо растворять не только катализатор, но и сам углеводород, а также его отделение от реакционной массы и возвращение в рецикл не должно вызывать трудностей. Хотя практика показывает, что окисление отдельных алкилароматических соединений в присутствии N-ГФИ можно осуществлять с высокими показателями процесса в гомогенной среде и без введения растворителя [15]. Второе направление базируется на использовании различных производных N-ГФИ, обладающих большей растворимостью в алкиларенах, нежели чем сам N-ГФИ. В работах [16–19] изучено влияние лиотропных производных N-ГФИ в полярном (ацетонитрил), неполярном (трет-бутилбензол) растворителях и в отсутствии растворителя. При этом удается повысить скорость окисления кумола по сравнению с использованием N-ГФИ. Несмотря на положительные результаты следует учитывать тот факт, что такие модифицированные катализаторы сложно синтезировать и они будут обладать высокой стоимостью. Кардинально другим направлением решения проблемы применения фталимидных катализаторов является разработка способа иммобилизации N-ГФИ на носителях для проведения процесса окисления в гетерогенной системе. Авторы статьи [20] указывают на то, что для создания гетерогенных фталимидных катализаторов, которые потенциально можно будет использовать в непрерывных поточных процессах, необходимо исследовать пути повышения и поддержания активности катализатора, стабильности опор, числа циклов его работы без дополнительной регенерации [20].
В настоящей работе представлены результаты наших исследований, направленных на изучение аэробного жидкофазного окисления циклогексана, циклогексанола и втор-бутилбензола с использованием N-гидроксифталимида и его аналогов.
Окисление циклогексана до циклогексанола и циклогексанона
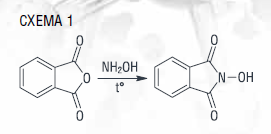
Получение циклогексанона (ЦГ-он) и циклогексанола (ЦГ-ол) – кетон-спиртового масла (КСМ) – является распространенным процессом в мировой химической промышленности. Основная часть потребления этих соединений приходится на производство адипиновой кислоты – полупродукта в синтезе капролактама, найлона-6 и найлона-6,6. Полиамидные (ПА) волокна и нити применяются в производстве текстиля, ковровых покрытий, промышленных нитей, используемых в свою очередь для изготовления шинного корда. Кордная нить – крупнейший и наиболее быстрорастущий сегмент рынка ПА6. Смола ПА6 также является основной для получения конструкционных пластиков, используемых для производства компонентов электронной и электрической техники, автомобильных деталей. В упаковочной отрасли находит применение ориентированная полиамидная пленка, изготовленная на основе ПА6. Также ЦГ-ол и ЦГ-он – растворители и стабилизаторы различной химической продукции [21, 22]. Промышленный синтез КСМ является двухстадийным: исходный циклогексан (ЦГ) окисляют до гидропероксида, затем последний в присутствии гомогенного кобальтового катализатора превращается в ЦГ-он и ЦГ-ол. Такой подход имеет следующие экономические и экологические недостатки: низкую конверсию 3–7 %, ввиду чего увеличиваются энергозатраты на отгонку непрореагировавшего сырья, невысокую селективность порядка 60–70 %, а также необходимость регенерации катализатора, вследствие чего образуются большие количества щелочных отходов. Таким образом, данная тема является полем активных исследований многих лабораторий, которыми были опробованы различные условия окисления циклогексана: использование гетерогенных катализаторов, содержащих благородные или переходные металлы, активированного угля; проведение реакции в присутствии растворителя или/и дорогостоящего окислителя. Все они не продемонстрировали значительного повышения эффективности и/или были дорогостоящими [23]. В наших исследованиях [24] был предложен альтернативный метод окисления циклогексана, отличающийся использованием органокатализа. Помимо этого, образующийся в ходе реакции ЦГ-ол в условиях проведения процесса окисляется в ЦГ-он, что обуславливает снижение количества побочных продуктов – адипиновой кислоты и ее эфиров. Предлагаемый способ осуществим без растворителя, а катализатор – N-ГФИ – является легкоотделяемым от реакционной массы и не требующим регенерации [24]. Данный катализатор может быть легко получен на основе доступного сырья (схема 1) и обладает высокой эффективностью в процессах окисления различных углеводородов [25–27].
Изучение жидкофазного окисления циклогексана в присутствии ацетата, стеарата и нафтената кобальта (II) показало, что процесс протекает с невысокими конверсией 3–4 % и селективностью 66–72 %. Тогда как окисление, катализируемое N-ГФИ, проходит с большей (примерно в два раза) конверсией и селективностью около 90 % (таблица 1).
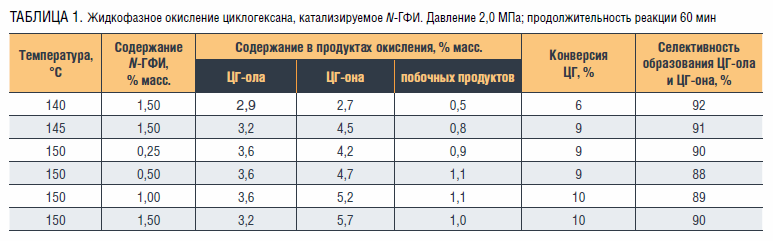
Наибольшее влияние на параметры окисления углеводорода оказывает температура: при ее повышении с 140 до 150 °С конверсия циклогексана возрастает с 6 до 10 %.
Использование N-ГФИ в сочетании с солями металлов переменной валентности является еще одним методом повышения эффективности окисления циклогексана [15, 16]. При использовании N-ГФИ с ацетатом кобальта при температуре 145 °C, давлении 2 МПа, за 1 час окисления достигается дополнительное увеличение конверсии циклогексана на 18–20 % и повышение селективности образования циклогексанола и циклогексанона с 88–90 % до
94–97 % (таблица 2). Это связано с синергетическим взаимодействием между двумя компонентами каталитической системы, вызываемым, по-видимому, образованием промежуточных комплексов с соединениями реакционной смеси. [17]. Установлено, что при повышении давления с 1,5 до 2,0 МПа при температуре 135 °С, времени реакции 60 мин и содержании в реакционной смеси 1,5 % масс. N-ГФИ и 0,15 % масс. ацетата кобальта (II) конверсия ЦГ увеличивалась с 6 до 9 %.
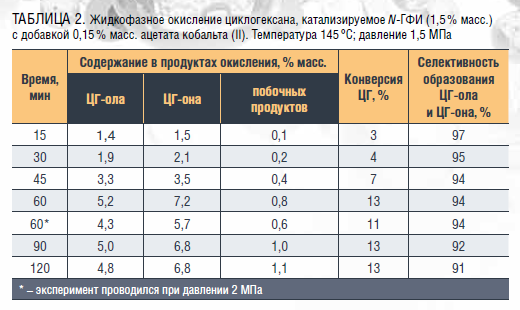
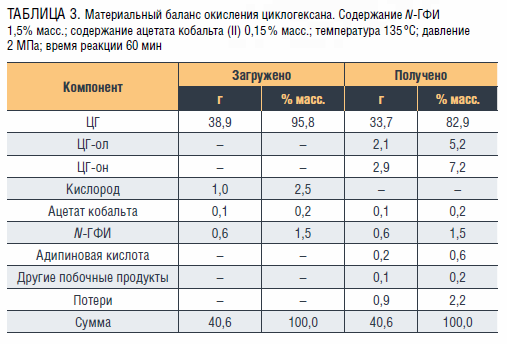
Установлено, что соотношение циклогексанона и циклогексанола в продуктах окисления составляет приблизительно 1,3–1,5:1. Наибольшие содержания спирта и кетона – 5,2 % и 7,2 % соответственно – достигаются в течение 1 ч окисления ЦГ при температуре 145 °C, давлении 1,5 МПа и массовом соотношении N-ГФИ : ацетат кобальта (II) 10 : 1. При указанных условиях селективность образования целевых продуктов составляет примерно 94 %. В таблице 3 представлен материальный баланс данного процесса.
Преимущество использования N-ГФИ в процессе окисления циклогексана заключается в том, что катализатор принимает участие в ряде последовательных реакций превращения циклогексанола в циклогексанон. Это, в свою очередь, уменьшает возможность последующего превращения циклогексанола в адипиновую кислоту и ее эфиры. Вследствие такой особенности процесса соотношение ЦГ-он : ЦГ-ол увеличивается с 1 : 1–2 до 1,5 : 1.
Механизм окисления циклогексана с использованием комплексного катализатора (N-ГФИ и ацетата кобальта II) может быть описан на основе известных концепций о механизмах каталитического окисления циклогексана с солями металлов переменной валентности [21] и углеводородов с фталимидными соединениями [3] (схема 2). Вероятно, процесс инициирования начинается с образования радикального комплекса Co(III)OO●, который взаимодействует с
N-ГФИ, образуя N-оксифталимидный радикал (N-ОФИР●).
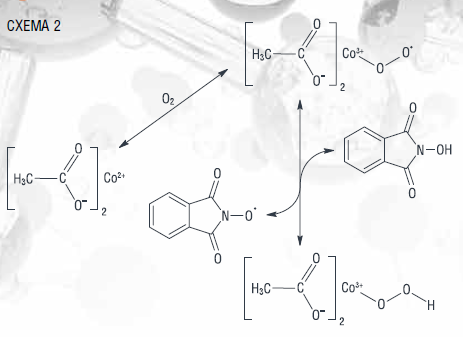
Превращение циклогексана в продукты окисления протекает по радикально-цепному механизму, включающему повторяющиеся циклы элементарных стадий. Особенностью этих стадий является то, что участвующие в реакциях свободные радикалы, включая N-ОФИР●, регенерируются в пределах каждого цикла [24, 28].
Высокая селективность (90–93 %) образования циклогексанола и циклогексанона, конверсия циклогексана (10–13 %), умеренная температура, доступный катализатор, позволяют говорить, что данный метод окисления циклогексана может представлять интерес для дальнейшего практического использования.
Окисление циклогексанола до циклогексанона
Циклогексанон относится к крупнотоннажным продуктам органического синтеза. Он широко используется лакокрасочными предприятиями как компонент для изготовления растворителей РЭ-7В, РЭ-11В. Также значительная его часть применяется в качестве сырья для производства пероксида циклогексанона, который входит в состав отвердителя ненасыщенных полиэфирных смол и различных лакокрасочных материалов в производстве стеклопластиков, полимербетонов, пуговиц, лакокрасочных покрытий и т.д. [29–31]. Кроме этого, данный кетон используется в фармацевтической и химической промышленности для изготовления медикаментов и инсектицидов.
Мировое производство циклогексанона составляет более 250 тысяч тонн в год, при ежегодном увеличении мощностей его производства. Быстрый рост потребления циклогексанона обуславливает актуальность создания новых высокоэффективных технологий его получения.
В промышленных масштабах циклогексанон в основном получают каталитическим дегидрированием циклогексанола (схема 3) при температуре
400–450 °С. Несмотря на достаточную селективность (80 %), этот процесс сопровождается образованием большого количества продуктов в виде смол и полимеров, что приводит к быстрой дезактивации катализатора. Другие известные способы получения данного соединения (окисление циклогексана (совместно получают и циклогексанол) или гидрирование фенола) также обладают рядом существенных недостатков [32].
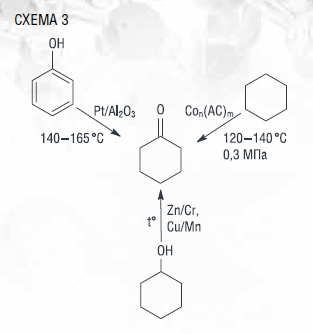
Альтернативой указанным выше способам может стать жидкофазное окисление циклогексанола. Существенным преимуществом этого метода получения циклогексанона является значительное снижение температуры (до 80–100 °С) и повышение селективности образования циклогексанона (более 95 %). В то же время сравнительно низкая конверсия циклогексанола сдерживает дальнейшую реализацию этого метода получения циклогексана. Повысить эффективность окисления циклогексанола в циклогексанон удалось за счет использования фталимидных катализаторов.
Изучение данного процесса проводили без применения растворителей, при температурах 90–120 °С и времени реакции 0,5–1,5 ч с использованием N-ГФИ в качестве катализатора [33].
Исследования показали, что существенное влияние на окисление ЦГ-ола оказывает количество катализатора, при повышении концентрации которого с 3,5 до 6,7 % мол. (время реакции – 1 ч, температура – 100 °С) содержание продукта реакции увеличивается с 9 до 32 % при достаточной высокой селективности 97–99 %. Также было изучено влияние температуры, продолжительности реакции и типа реактора, результаты представлены в таблице 4. Было установлено, что фталимидный катализатор легко возвращать в рецикл, его активность при этом остается прежней в пределах 3–5 повторных использований
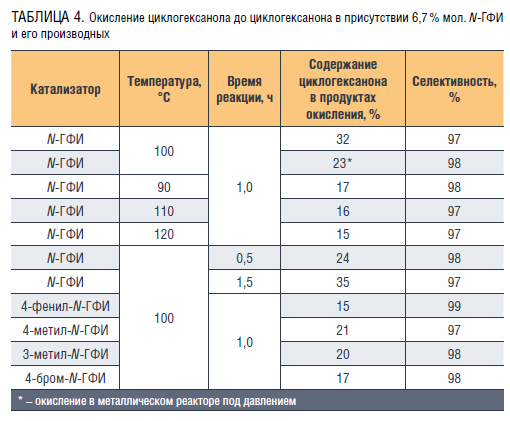
Наибольшее содержание циклогексанона в продуктах реакции окисления – порядка 30–32 % – достигается при температуре 100 °C в течение 1 часа реакции. Изменение температуры выше или ниже 100 °C не приводит к желательным результатам. При проведении окисления циклогексанола в атмосфере воздуха в металлическом реакторе, используя установку типа «УОСУГ», содержание циклогексанона в продуктах окисления составило 23 %, а селективность его образования достигла 98 % при давлении 5 атмосфер при температуре 100 °C.
Известно, что улучшение процесса окисления углеводородов может быть достигнуто с использованием фталимидных катализаторов, содержащих электроноакцепторные или электронодонорные заместители [34]. Этот путь не оказался эффективным при окислении циклогексанола (см. таблицу 4).
Другим методом для повышения эффективности окисления является использование каталитической системы N-ГФИ – соли металлов переменной валентности, о чем упоминалось ранее в настоящей работе. Исследование влияния состава каталитической системы на окисление циклогексанола показало, что совместное использование N-ГФИ с ацетатом или стеаратом двухвалентного кобальта позволяет достичь 45–50 % конверсии циклогексанола, при сохранении высокой селективности (таблица 5).
Полученные данные позволяют сделать вывод о том, что увеличение количества N-ГФИ оказывает наиболее существенное влияние на эффективность процесса. Приемлемым временем реакции является 1 час ввиду того, что при окислении в течение большего времени циклогексанон окисляется далее до адипиновой кислоты и др. соединений, а в обратном случае – при окислении в течение получаса – целевое вещество не успевает накопиться в реакционной массе в достаточном количестве. В качестве сокатализатора более предпочтительным является использование ацетата кобальта (II).
Механизм окисления циклогексанола заключается в том, что
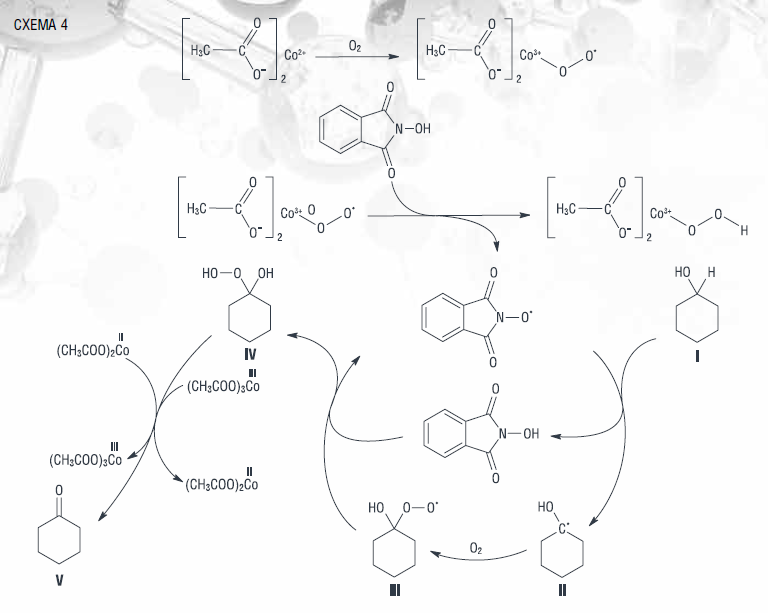
N-оксифталимидный радикал, образующийся при инициировании солью, отрывает атом водорода от циклогексанола (I) с образованием α-гидроксикарборадикала (II), который, реагируя с кислородом, превращается в α-гидрокси-α-пероксирадикал (III). Последний трансформируется в циклогексанон (V) через образование промежуточного α-гидроксигидропероксида (IV) (схема 4) [3, 33].
Гидропероксидный подход к получению кислородсодержащих соединений
Развитие технологии крупнотоннажных нефтехимических производств – одна из важнейших задач современной технологии органических веществ, и развитие совмещенных синтезов как ее часть позволяет в комплексе решать не только проблему самого синтеза, но и его экономическую и экологическую составляющие, которые играют решающую роль в конкурентоспособности продукта в современном быстроразвивающемся мире. К таким технологиям можно отнести синтез различных веществ на основе гидропероксидного окисления алкилароматических углеводородов.
Гидропероксидный подход к получению кислородсодержащих соединений существует уже около 80 лет. За свою почти вековую историю этот метод был достаточно хорошо изучен, реализован в промышленности и до сих пор применяется для синтеза фенолов, кетонов и других ценных кислородсодержащих соединений [35]. Остаются актуальными новые исследования с целью интенсификации процесса получения востребованных в ближайшем будущем соединений и модернизации существующих производств ввиду изменения конъюнктуры рынка продуктов основного органического и нефтехимического синтеза.
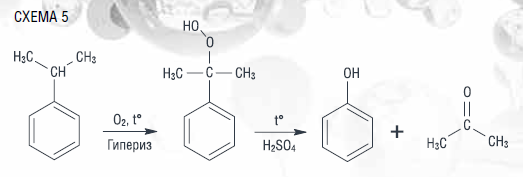
Кумольная технология (или процесс Хока) широко распространена в мировой химической промышленности для получения фенола и ацетона. На производстве изопропилбензол (ИПБ, кумол) окисляют до гидропероксида в присутствии инициатора гипериза (гидропероксида кумола) при температурах 100–120 °C. Гидропероксид кумола прекращает свое существование в установках с отводом тепла испаряющимся ацетоном или установках проточно-циркуляционного типа с образованием смеси исходного углеводорода, фенола и ацетона, которые далее разделяют, получая нужные продукты (схема 5).
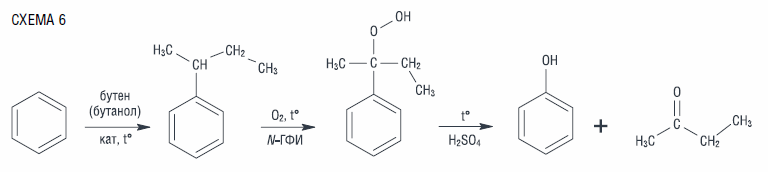
Вследствие экономических факторов, развития и изменений рынка химической продукции, ацетон становится менее востребованным веществом, следовательно, и технология становится менее рентабельной. Поэтому были предприняты попытки усовершенствовать гидропероксидный способ получения фенола. Компанией «Illa International» проводились исследования окисления втор-бутилбензола (ВББ), но окисление смеси последнего с кумолом не оказалось достаточно эффективным, хоть и позволило получать фенол, ацетон и метилэтилкетон [36]. Окисление же индивидуального втор-бутилбензола (схема 6) имеет перспективы реализации в промышленности ввиду доступности бутенов, востребованности метилэтилкетона во многих отраслях промышленности:
N-ГФИ при окислении ИПБ показал отличительные каталитические свойства – содержание ГП ИПБ достигает 34 % масс. при температуре 120 °C за 2 ч реакции, что значительно выше показателей существующей кумольной технологии. Это обуславливает возможность применения катализатора в исследованиях аэробного окисления других алкилароматических соединений. Нами было изучено окисление обсуждаемого углеводорода в присутствии N-ГФИ (рисунок 1 а, б). Содержание ГП ВББ в продуктах реакции достигает 35 % при температуре 140 °C за 1 ч окисления в присутствии 2 % масс. N-ГФИ.
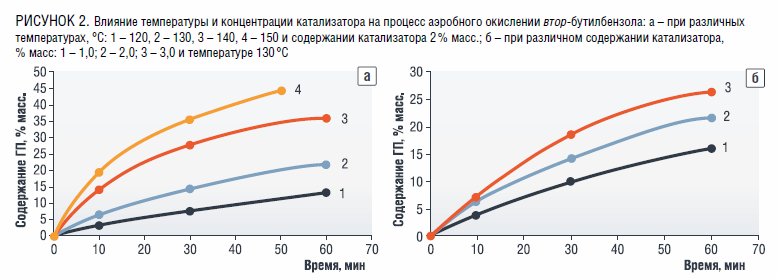
Увеличение же загрузки катализатора до 4 % масс. позволяет получать
до 27 % ГП ВББ при температуре 130 °C и времени реакции 1 ч. Установлено, что селективность процесса падает с 97–98 до 92–94 % при повышении температуры реакции до 140 °C и выше, а также менее значительно уменьшается при увеличении времени реакции и концентрации катализатора.
Также было изучено влияние структуры фталимидных катализаторов на основные показатели процесса окисления ВББ до соответствующего гидропероксида. Результаты исследований представлены в таблице 6.
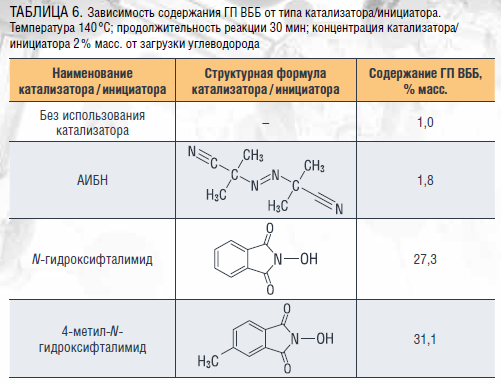
Наряду с изучением окисления втор-бутилбензола было проведено комплексное исследование аэробного жидкофазного окисления алкиларенов – кумола, циклогексилбензола и их алкильных производных – до гидропероксидов в присутствии фталимидных соединений, доказана их высокая каталитическая активность и в этих процессах. Найдены условия, позволяющие получать гидропероксиды алкилароматических углеводородов с высокими выходом и селективностью. Полученные результаты открывают путь для будущих исследований, направленных на усовершенствование действующих производств крупнотоннажных продуктов органического синтеза [37, 38]
Механизм окисления алкилароматических углеводородов до гидропероксидов в присутствии N-гидроксифталимида
С научной точки зрения, а также для проектирования химико-технологического процесса жидкофазного аэробного окисления алкилароматических углеводородов в присутствии фталимидных катализаторов, его аппаратурного оформления и расчета реакционного узла немаловажное значение имеет изучение механизма.
Известно, что жидкофазное окисление алкиларенов протекает по радикально-цепному механизму через элементарные стадии зарождения, роста и обрыва цепи [39, 40].
Схему окислительных превращений углеводородов можно представить в виде следующих элементарных актов реакций (I)–(VII):
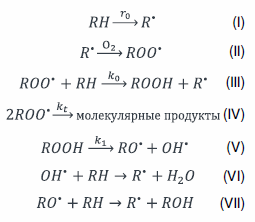
При введении в систему N-гидроксифталимида его роль заключается в следующем: инициирование и ускорение процесса окисления, повышение селективности образования ГП. При этом он не участвует в процессах, приводящих к дестабилизации гидропероксида, что позволяет накапливать высокие концентрации последнего при окислении. Если же рассматривать катализ с использованием солей металлов переменной валентности процесс аэробного окисления углеводородов протекает с образованием в качестве промежуточного продукта соответствующего гидропероксида. Металлы переменной валентности и их производные с одной стороны ускоряют процесс окисления, а с другой приводят к нецелевому разложению гидропероксида с образованием спиртов, кетонов, воды и кислорода.
Подробнее остановимся на механизме аэробного окисления углеводородов в присутствии фталимидных соединений. Известно, что реакция зарождения цепи, связанная с возникновением в реакционной среде пероксидных радикалов, как правило, протекает с индукционным периодом, для сокращения которого вводят различного рода инициаторы. Исследования показывают, что окисление алкиларенов кислородом инициируется N-гидроксифталимидом, но он не является «классическим» инициатором. Первоначально, со скоростью прямо пропорциональной концентрации N-ГФИ, устанавливается стационарная концентрация N-оксифталимидных радикалов. Скорость инициирования всего процесса определяется реакцией N-ОФИР● с углеводородом с образованием алкильных радикалов (R●) и далее – пероксильных радикалов (ROO●). Путь инициирования процесса через радикальный распад гидропероксида ограничивается взаимодействием образующихся радикалов RO● и HO● с катализатором и образованием N-оксифталимидного радикала.
Необходимо отметить, что N-оксифталимидные радикалы распадаются гораздо медленнее, чем пероксильные радикалы, что делает их более эффективными переносчиками цепи. Введение в реакционный раствор
N-гидроксиимидов уменьшает стационарные концентрации радикалов ROO● и RO●, тем самым уменьшая возможность их участия в образовании продуктов распада гидропероксида. Таким образом, можно сделать вывод о том, что данные соединения катализируют реакцию продолжения цепи при снижении скорости квадратичного обрыва.
Процесс свободно-радикального окисления углеводородов в присутствии N-гидроксифталимида можно представить следующим образом (схема 7):
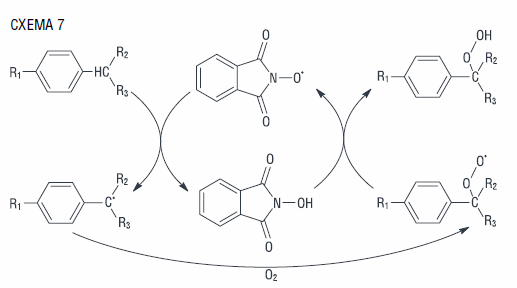
Высокое значение константы скорости взаимодействия N-оксифталимидного радикала с RH позволяет предположить наличие туннельного переноса атома водорода от углеводорода на радикал катализатора [41].
Описанные выше особенности придают N-гидроксифталимиду и родственным соединениям уникальные каталитические свойства в радикальных процессах.
Заключение
Применение фталимидных органических соединений в качестве катализаторов является объектом интереса многих исследователей при поиске новых путей осуществления селективных химических процессов. В настоящее время все с большей уверенностью можно говорить, что достаточная научно-теоретическая база для практического освоения и скорейшего внедрения технологии органокатализа сформирована и обладает социально-экономическим потенциалом, способным определить конкурентоспособность страны. Технология, затрагивающая многочисленные сектора химической промышленности и нефтепереработки, обладает ключевым значением для различных сфер жизнедеятельности нашего общества.
Комплекс проведенных нами исследований подтвердил высокую эффективность использования N-гидроксифталимида и его производных при жидкофазном аэробном окислении циклогексана, циклогексанола и широкого ряда алкилароматических углеводородов до соответствующих кислородсодержащих органических соединений, являющихся ценными продуктами основного органического синтеза. Установлено, что данные каталитические системы повышают конверсию исходного сырья, скорость и селективность образования целевых продуктов и при всем этом не участвуют в разложении гидропероксида, т.е. укрепление оксидатов в их присутствии будет является технологически приемлемым. Обеспечение высокой конверсии углеводорода за один подход может значительно сократить энергоемкость соответствующих производств, так как именно на выделение и рецикл непревращенного сырья приходится основная часть энергозатрат.
Все изложенное выше подтверждает перспективность фталимидных катализаторов и их уникальные свойства, которые они проявляют в процессах окислительных превращений углеводородного сырья.
Литература
1. L. Cohn. Phtalylhydroxylamin: Ueberführung der Phtalsäure in Salicylsäure // Justus Liebig’s Annalen Der Chemie – 1880 – Vol. 205 – P. 295–314.
2. E. Grochkowski, T. Boleslawska, J. Jurczak. Synthesis new strategy for alkane oxidation with O2 using N-hydroxyphthalimide (NHPI) as a radical // Catalysis Surveys from Asia – 1977 – 718 р.
3. Y. Ishii, S. Sakaguchhi, T. Iwahama. Innovation of hydrocarbon oxidation with molecular oxygen and related reactions // Adv. Synth. Catal. – 2001 – Vol. 343 (5) – P. 393–427.
4. M. Masui. N-Hydroxyphtalimide as an mediator in the anodic oxidation of organic compounds // Resent Advances in Electro organic Synthesis. New York: Marcel Dekker – 1989 – P. 137–144.
5. F. Recupero, C. Punta. Free Radical Functionalization of Organic Compounds Catalyzed by N-Hydroxyphthalimide // Chemical Reviews – 2007 – Vol. 107(9) – P. 3800–3842. doi:10.1021/cr040170k.
6. L. Melone, C. Punta. N-Hydroxyphthalimide (NHPI)-Organocatalyzed Aerobic Oxidations: Advantages, Limits, and Industrial Perspectives. Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives – 2016 – P. 253–265. doi:10.1002/9783527690121.ch16.
7. M.M. Al-Taher, C. Kalamaras, M.A. Alqahtani, F.S. Alomar. Aerobic oxidation of hydrocarbons using N-hydroxyphthalimide (NHPI) catalysts for cetane enhancement // Fuel – 2022 – Vol. 324, Part B., 124563
8. И.В. Карпенко, А.А. Новохатько, М.А. Компанец, Е.В. Новикова, О.В. Кущ, Л.И. Опейда, Е.В. Карпенко. Влияние структуры N-гидроксифталимидов на их биологическую активность // Материалы XIV Междунар. науч.-практ. конф., Минск: БГУ – 2018 – С. 89–91.
9. Andy N.S. Shamaya, Oday H.R. Al-Jeilawi, Noor Ali Khudhair. Novel Synthesis of Some N-Hydroxy Phthalimide Derivatives with Investigation of Its Corrosion Inhibition for Carbon Steel in HCl Solution // Chem. Methodol. – 2021 – Vol. 5, I. 1 – P. 331–340.
10. Alya M. Al-Etaibi, Nouria A. Al-Awadi, Maher R. Ibrahim, Yehia A. Ibrahim. Gas-phase pyrolysis of N-alkoxyphthalimides to functionally substituted aldehydes: kinetic and mechanistic study // ARKIVOC – 2010 – Vol. 2010 (x): General Papers – P. 149–151.
11. D.L. Golden, S.-E Suh, S.S. Stahl. Radical C(sp3)–H functionalization and cross-coupling reactions // Nature Reviews Chemistry – 2022 – Vol. 6 – P. 405–427.
12. H.-M. Huang, M. Koy, E. Serrano, P.M. Pflüger, J.L. Schwarz, F. Glorius. Catalytic radical generation of π-allylpalladium complexes // Nature Catalysis – 2020 – Vol. 3 (4) – P. 393–400. doi:10.1038/s41929-020-0434-0.
13. L. Melone, S. Prosperini, G. Ercole, N. Pastori, C. Punta. Is it possible to implement N-hydroxyphthalimide homogeneous catalysis for industrial applications? A case study of cumene aerobic oxidation // Journal of Chemical Technology & Biotechnology – 2013 – Vol. 89 (9) – P. 1370–1378. doi:10.1002/jctb.4213
14. Е.А. Курганова, Э.М. Дахнави, Г.Н. Кошель. Окисление изопропилбензола до гидропероксида в присутствии N-гидроксифталимида // Нефтехимия. – 2017 – Т. 57, № 2 – С. 204–208.
15. R. Amorati, M. Lucarini, V. Mugnaini, G. F. Pedulli, F. Minisci, F. Recupero, F. Fontana, P. Astolfi, L. Greci. Hydroxylamines as Oxidation Catalysts: Thermochemical and Kinetic Studies // Journal of Organic Chemistry. – 2003 – V. 68, № 5 – P. 1747–1754.
16. K. Kasperczyk, B. Orlinska, J. Zawadiak. Aerobic oxidation of cumene catalysed by 4-alkyloxycarbonyl-N-hydroxyphthalimide // Central European Journal of Chemistry. – 2014 – V. 12, № 11 – P. 1176–1182.
17. M. Caruso, S. Navalón, M. Cametti, A. Dhakshinamoorthy, C. Punta, H. García. Challenges and opportunities for N-hydroxyphthalimide supported over heterogeneous solids for aerobic oxidations // Coordination Chemistry Reviews – 2023 – Vol. 486, 215141.
18. Е.Л. Красных, С.В. Леванова, А.Б. Соколов, И.Л. Глазко. Получение ароматических углеводородов из отходов производства капролактама // Хим. пром. сегодня – 2004 – С. 27–31.
19. M. Sadiq, M. Khan, M. Numan, R. Aman, S. Hussain, M. Sohail Ahmad, S. Sadiq, M. Abid Zia, H. Ur Rashid, R. Ali. Tuning of Activated Carbon for Solvent-Free Oxidation of Cyclohexane // Journal of Chemistry – 2017 – Vol. 2017 – P. 1–8. doi:10.1155/2017/5732761.
20. А.С. Фролов, Е.А. Курганова, Е.М. Яркина, Н.В. Лебедева, Г.Н. Кошель, А.С. Каленова. Интенсификация процесса жидкофазного окисления циклогексана // Тонкие химические технологии – 2018 – Т. 13, № 4 – С. 50–57.
21. Е.А. Курганова, Г.Н. Кошель. Жидкофазное окисление алкилароматических углеводородов и их циклогексильных производных до гидропероксидов в присутствии фталимидных катализаторов // Российский химический журнал – 2014 – Том LVIII, 3–4 – С. 91–102.
22. И.А. Опейда, А.Л. Плехов, О.В. Кущ, А.Г. Матвиенко. Комплексы N-гидроксифталимида и ацетата кобальта (II) в реакциях окисления алкиларенов молекулярным кислородом // Журн. физ. xимии – 2011 – Т. 85. № 7 – С. 1223–1228.
23. Пат. РФ 2046793C1. Стабильный раствор пероксида циклогексанона, не кристаллизующийся до температуры минус 20 °C и способ его получения: заявл. 13.08.1993; опубл. 27.10.1995.
24. J. Bińczak, A. Szelwicka, A. Siewniak, K. Dziuba, A. Chrobok. Oxidation of Cyclohexanone with Peracids – A Straight Path to the Synthesis of ε-Caprolactone Oligomers // Materials – 2022 – Vol. 15 (19), 6608.
25. K. Xiangqian, G. Yutong, M. Shanjun, W. Yong. Selective Hydrogenation of Phenol // ChemNanoMat – 2018 – Vol. 4 – P. 1–20. 10.1002/cnma.201800031.
26. Е.А. Курганова, К.А. Пуркарьян, А.С. Фролов, Н.В. Лебедева, Кошель Г.Н. Аэробное окисление циклогексанола в циклогексанон, катализируемое
N-гидроксифталимидом // Вестник Казанского технологического университета – 2016 – Том 19, № 2 – С. 53–56.
27. В.М. Закошанский Альтернативные технологии получения фенола // Ж. Рос. хим. об-ва им. Д.И. Менделеева – 2008 – Т. LII, № 4 – P. 53–71.
28. Е.А. Курганова, В.С. Кабанова, А.С. Фролов, Г.Н. Кошель, А.А. Смурова, Е.И. Баёв. Гидропероксидный способ синтеза фенола и его алкильных производных совместно с кетонами алифатического и алициклического ряда // Neftegaz.RU – 2023 – № 5 – С. 90–95.
29. Семёнов Н.Н. Цепные реакции // Госхимиздат, 1940 – 268 с.
30. V.N. Sapunov, E.A. Kurganova, G.N. Koshel. Kinetics and Mechanism of Cumene Oxidation Initiated by N-Hydroxyphthalimide // International Journal of Chemical Kinetics – 2018 – V.50 (1) – P. 3–14.



